
|
Molinspiration Services FAQ |
Free Molinspiration property calculation service is up since January 2002 and since then become already well established resource for the internet chemistry community (at least according to the usage numbers, reaching currently about 100,000 calculations per month!).
We would like to thank all users who sent us questions, comments and suggestions. Thank to this feedback we could implement already several improvements, and we will try do so also in the future.
Below you can find a short collection of the most common questions we are getting, together with the answers. If your topic is not covered, do not hesitate to send us mail to info [at] molinspiration.com.
May I use calculated data in a publication?
Yes, you can use calculated properties freely for any non-commercial activities (publications, lectures, research reports, web documents). You can use also screenshots of our tools in your presentations (thanks to people who did it already). We would like to ask you, however, to acknowledge the Molinspiration Property Calculation Service, and mention our web address www.molinspiration.com (including adding a link to it in your on-line documents).
See also a growing list of publications citing Molinspiration software (be so kind and send us update if your work is not mentioned).
For more information check our Terms of Service document.
How should I cite the Molinspiration web services in my publication?
Use the following citation:
Molinspiration Cheminformatics free web services, https://www.molinspiration.com, Slovensky Grob, Slovakia
or something similar as required by the respective journal citation policy
Are your logP values better than CLOGP?
CLOGP is still considered to be a "gold standard" in logP prediction. We hope, however, that our results are of comparable quality with the CLOGP ones, and useful in drug design and QSAR projects. Several Molinspiration licensees reported very good correlation of our logP values with their in-house data and with various experimental drug properties. And the statistical results (r2 = 0.944, r = 0.972, stdev = 0.428 for 12'202 molecules) also prove the very good quality of the Molinspiration miLogP model.
May I use your service in my medicinal chemistry / cheminformatics / QSAR class?
Yes, feel free to do so. Our property prediction service are used already in such classes at several leading universities. If you want to use our calculation engines locally for educational purposes, contact us please for special arrangement.
I need help with my QSAR / diversity / ... analysis. I need to design a combichem library focused on GSK-3 kinase inhibitors.
Molinspiration provides consultancy in the area of cheminformatics, QSAR, virtual screening, and drug design. Contact us please.
Why the number of calculations is limited to 1000 per month?
First of all, to keep the load on our server low, to guarantee a good performance for all users, and secondly to prevent some "experts" to use automatic scripts to calculate (and later sell) properties for large molecular databases. If you need more data for your research project, let us know, and we may consider to increase the limit for your IP address (or even to calculate the data for you, if you will send us molecule SMILESes). If you need to calculate data for large number of molecules on a regular basis, purchase our property calculation engine.
Why is the SMILES input not available?
Sorry for the inconvenience, but we had to remove the SMILES input box for the following reason: Several criminals, completely ignoring our user policy, massively misused the SMILES input, calculating data for a huge number of molecules by automatic scripts. This led to the high load on our servers, blocking other users and crashes. Our options were either to completely stop the free services or to remove the SMILES input. You can now input your molecules by the JSME editor on the input page. If you need to calculate properties for a large number of molecules encoded as SMILES you may purchase our software for local use.
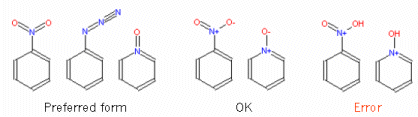
How should I correctly input nitro derivatives and N-oxides?
Input nitro groups, N-oxides and related structures with pentavalent nitrogen as shown below. You may use the "y" keyboard shortcut in JSME to input the nitro group. The form with a polar bond is also accepted. Do not enter this type of molecules with the -OH bond (this is a quite common error according to our logs) !
May I install this property calculation locally on my machine?
Yes, we sell the property calculation engine which powers this service. You can use it as a web engine (we can offer help with the web deployment) or use it in a batch mode to calcule properties for hundreds of thousands molecules. You are not allowed, however, to provide property calculation services publicly on the internet.
Alternatively, you may consider purchasing Molinspiration Property Calculator which allows easy interactive calculation of molecular properties on your desktop.
What about confidentiality of structures I am submitting to your servers?
All Molinspiration services use secure https internet connection. The processed structures are logged on our servers and logs are periodically checked to identify possible errors or issues. We have, of course, no information about identity of users (with exception of their IP addresses) If you need to process highly confidential structures, you may contact us to arrange free local evaluation of our tools, and if you are satisifed you may consider purchasing the Molinspiration software.
Where can I find the calculations of Bioactivity Scores?
Unfortunately, we have had to remove the calculations of Bioactivity Scores for the time being. Maintaining this service is highly resource-intensive, as it requires frequent updates with the latest published bioactivity data and thorough testing of the models.
Regrettably, we currently lack the resources to support these ongoing efforts. If you find this service valuable and are interested in sponsoring its continued maintenance, please don't hesitate to contact us.
December 2024, your Molinspiration team.